

Vol. 19 - Num. 74
Notas clínicas
Síndrome de Prader-Willi: diagnóstico en el periodo neonatal
Beatriz Cano del Águilaa, Roberto Ortiz Movillab, Gema Iglesias Escalerac, Itziar Martínez Badásc
aMIR-Pediatría. Hospital Universitario Puerta de Hierro. Majadahonda. Madrid. España.
bUnidad de Neonatología. Servicio de Pediatría. Hospital Universitario Puerta de Hierro. Majadahonda. Madrid. España.
cServicio de Pediatría. Unidad de Neonatología. Hospital Universitario Puerta de Hierro. Majadahonda. Madrid. España.
Correspondencia: B Cano. Correo electrónico: bea.c028@gmail.com
Cómo citar este artículo: Cano del Águila B, Ortiz Movilla R, Iglesias Escalera G, Martínez Badás I. Síndrome de Prader-Willi: diagnóstico en el periodo neonatal. Rev Pediatr Aten Primaria. 2017;19:151-6.
Publicado en Internet: 05-06-2017 - Número de visitas: 25534
Resumen
El síndrome de Prader-Willi debe formar parte del diagnóstico diferencial en los casos de hipotonía de presentación neonatal de origen central, especialmente cuando se acompaña de otras características clínicas típicas de la enfermedad. La importancia de su diagnóstico precoz, ya sea en el ámbito hospitalario o desde las consultas de Atención Primaria, reside tanto en la necesidad de instaurar una adecuada estimulación y fisioterapia por parte de los Servicios de Atención Temprana como de un correcto soporte nutricional en los que debutan con trastorno importante de succión. Así mismo es importante ofrecer un correcto consejo genético para planificar futuras gestaciones. Presentamos el caso clínico de un recién nacido con hipotonía de presentación neonatal junto con criptorquidia bilateral y fenotipo peculiar, en el que se confirmó, mediante estudio genético, el diagnóstico de sospecha de síndrome de Prader-Willi.
Palabras clave
● Criptorquidia ● Hipotonía neonatal ● Síndrome de Prader-WilliINTRODUCCIÓN
El síndrome de Prader-Willi (SPW) es una alteración genética de escasa incidencia, debida a la falta de expresión de genes situados en el brazo largo del cromosoma 15, de herencia paterna1,2. Las manifestaciones clínicas varían con la edad, y a largo plazo es una de las causas más frecuentes de obesidad de origen genético. Sin embargo, durante la época neonatal destacan la hipotonía generalizada y los problemas de alimentación en los recién nacidos afectos. El diagnóstico de sospecha se debe realizar con test genéticos específicos que confirmen esta entidad y que nos ayudarán a establecer el riesgo de recurrencia en la futura descendencia.
CASO CLÍNICO
Varón de 14 días de vida que acude a consultas externas de Neurología y Endocrinología para la valoración de una hipotonía marcada desde el nacimiento y criptorquidia bilateral.
Nace en otro centro, tras gestación espontánea, controlada, de curso normal hasta las 33 semanas, cuando inicia estancamiento ponderal. Líquido amniótico normal, sin polihidramnios y movimientos fetales normales desde el cuarto mes. El parto fue por cesárea a las 36 + 6 semanas, por riesgo de pérdida de bienestar fetal. Test de Apgar al minuto/cinco minutos: 8/9. Biometría de recién nacido: peso 2130 g (p5), longitud 45,5 cm (p7), perímetro cefálico 33 cm (p44).
No presentaba antecedentes familiares de interés, siendo el segundo hijo de pareja no consanguínea, con una hija previa sana. Ingresa en la Unidad de Neonatología de su hospital de referencia durante los primeros tres días de vida, con los diagnósticos de recién nacido de bajo peso, hipoglucemia precoz asintomática (que requirió corrección intravenosa las primeras horas de vida, por dificultad para la alimentación enteral) e hipotonía con fenotipo peculiar, realizándose cribado analítico básico (normal) y ecografías intracraneal, abdominal y testicular, en donde se observa criptorquidia bilateral; el resto de pruebas de imagen son normales. Se extrae asimismo una muestra para estudio genético (cariotipo) y es dado de alta a los tres días de vida, con buena evolución clínica, alimentación con lactancia materna suplementada y recomendación de revisión en centro de referencia.
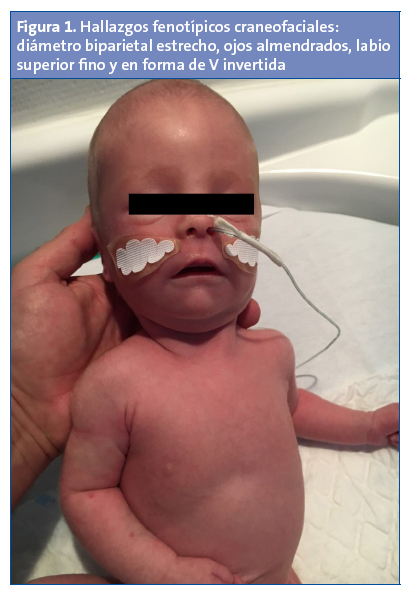
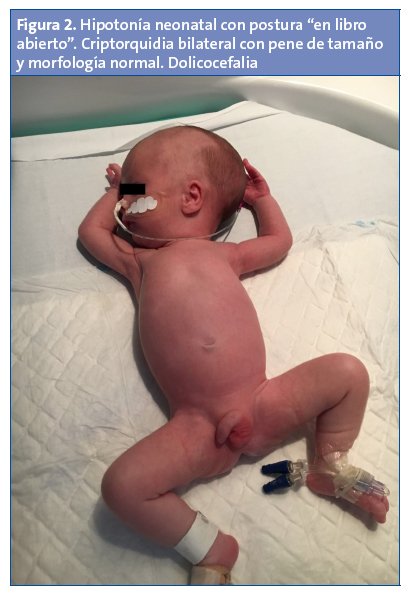
En la exploración física en nuestras consultas se observa un fenotipo peculiar con dolicocefalia, ojos almendrados, orejas pequeñas de implantación baja y boca pequeña con paladar ojival. Presenta asimismo criptorquidia bilateral con bolsas escrotales hipoplásicas, con pene de tamaño y morfología normal (Figs. 1 y 2). En la exploración neurológica se evidencia una llamativa hipotonía generalizada de predominio axial, con postura de libro abierto en decúbito y motilidad general escasa, aunque con posibilidad de elevar miembros superiores e inferiores contra la gravedad, hipomotilidad facial, succión pobre, con llanto escaso. No presenta asimetrías faciales, tiene adecuados movimientos oculares, reflejos osteotendinosos presentes aunque levemente disminuidos, reflejo del Moro simétrico y normal. El resto de reflejos del recién nacido fueron normales, salvo el reflejo de succión que era muy débil. El resto de la exploración neurológica fue normal. Caderas laxas, sin otros signos de inestabilidad.
Debido a la marcada hipotonía y la pobre succión que condicionaba una importante dificultad para la alimentación, presentaba una ganancia ponderal muy pobre, por lo que ingresa en Neonatología para completar el estudio y realizar soporte nutricional con sonda nasogástrica. Durante su ingreso en la unidad neonatal precisa inicialmente soporte respiratorio con gafas nasales de alto flujo (4-5 l/min, fracción inspiratoria de oxígeno en el aire inspirado máxima del 30%) por saturaciones de oxígeno por debajo de la normalidad e hipercapnia secundaria a hipoventilación por hipotonía central. El cuadro respiratorio se resolvió a los cinco días del ingreso. Precisa asimismo suplemento de tomas por sonda nasogástrica con fórmula hipercalórica, con paso progresivo a vía oral, mostrando con el paso de los días una adecuada, aunque lenta, coordinación succión-deglución, con curva ponderal también lentamente ascendente. Se inician medidas de rehabilitación y estimulación precoz durante su ingreso, mostrando mejoría en la motilidad general, aunque persiste una hipotonía axial marcada. Se puede retirar la sonda nasogástrica a los 35 días de vida, manteniendo posteriormente adecuada ganancia ponderal con alimentación enteral exclusiva, siendo dado de alta dos días después. El resto de pruebas analíticas y ecográficas (abdominal, craneal, ecocardiografía, caderas) realizadas durante su hospitalización no mostraron resultados relevantes, salvo la confirmación de criptorquidia bilateral con ambos testes en el tercio proximal de sus respectivos canales inguinales.
Durante su estancia en la Unidad Neonatal se recibieron resultados del estudio genético realizado en su hospital de origen, que confirmó un cariotipo 45,XY, cromosoma 15, con deleción de la región Prader-Willi/Angelman, producida por una translocación no recíproca entre los cromosomas 15 y 16, de novo, con ausencia de alteración en el cariotipo de ambos progenitores. Con el diagnóstico de SPW, se da el alta y se indica seguimiento ambulatorio multidisciplinar en consultas externas de Neonatología, Gastroenterología, Neurología y Endocrinología Infantil.
DISCUSIÓN
El síndrome de Prader-Willi es un trastorno poco común (prevalencia 1/15 000-1/30 000 recién nacidos vivos), a consecuencia de la ausencia física o funcional de genes que se expresan solo a partir del cromosoma 15 de herencia paterna1,2. Se han descrito tres mecanismos genéticos que resultan en la afectación de dichos genes, como la microdeleción de un fragmento de ADN en el cromosoma 15 paterno (65-70% de los casos), la disomía uniparental materna (cuando ambos cromosomas 15 son de origen materno, responsable de un 20-30% de los casos) y menos frecuente (5%) por defectos en la impronta genómica, de manera que los genes paternos están presentes, pero “silenciados”3,4.
Las manifestaciones clínicas varían con la edad. Durante el periodo prenatal pueden presentar una posición fetal anómala, polihidramnios y movimientos fetales disminuidos2. En la época neonatal llama la atención una hipotonía marcada, de origen central, de predominio axial, con movimientos espontáneos disminuidos, posición en libro abierto y letargia, con llanto débil. El reflejo de succión esta disminuido, lo que conlleva una importante dificultad para la alimentación, y como consecuencia un estancamiento ponderal, precisando en la mayoría de los casos soporte nutricional con colocación de sonda nasogástrica2. Hoy en día la gastrostomía es excepcional, ya que la hipotonía disminuye parcialmente con la edad, por lo que mejora la alimentación. El fenotipo en este periodo también muestra una serie de rasgos craneofaciales característicos como dolicocefalia, diámetro bifrontal estrecho y boca pequeña con labio superior fino, y otros hallazgos como manos y pies pequeño y criptorquidia. Existe asimismo un mayor riesgo de displasia del desarrollo de caderas2. En nuestro paciente, los hallazgos fenotípicos (rasgos craneofaciales, criptorquidia, hipotonía marcada…) nos hicieron sospechar esta enfermedad, aunque en el periodo neonatal otras enfermedades neuromusculares (atrofia muscular espinal, distrofia miotónica…), genéticas o metabólicas pueden producir un cuadro similar y deben ser tenidas en cuenta a la hora de realizar el correspondiente diagnóstico diferencial.
Este síndrome, primera causa de obesidad de origen genético, condiciona una disfunción hipotalámica que dará lugar a varias de las manifestaciones clínicas a largo plazo características de la enfermedad, consecuencia tanto de la ausencia de sensación de saciedad como de una disminución global del metabolismo basal y del requerimiento calórico total2. La falta de saciedad comienza en torno a los 1-4 años y condiciona la aparición de conductas anormales en relación con la comida (búsqueda de alimentos, robo de comida, atracones…). Por tanto, ya desde la infancia, estos niños precisan de un control muy estricto de la ingesta1,2. Es importante concienciar a las familias de la necesidad de este control para evitar el desarrollo de obesidad mórbida, ya que esta supone la principal causa de morbilidad y mortalidad de estos pacientes1. Se ha observado también una disminución de los niveles séricos de la hormona del crecimiento (GH) y el factor de crecimiento similar a la insulina tipo 1 (IGF-1) en estos pacientes, dando lugar a una alteración de la composición corporal, con aumento de la masa grasa y disminución de la masa muscular, y baja estatura. El tratamiento con GH ha demostrado una mejora en el crecimiento lineal, la composición corporal y la densidad ósea a largo plazo.
Otra característica distintiva de este síndrome es el hipogonadismo. Este es de origen hipotalámico y afecta a ambos sexos, de modo que en el varón encontraremos un escroto hipoplásico, poco pigmentado, y criptorquidia unilateral o bilateral en un alto porcentaje de recién nacidos (80-90%). El pene puede ser pequeño. En la mujer habrá labios menores, mayores y clítoris hipoplásicos. El inicio del desarrollo puberal estará retrasado y será incompleto2.
Desde el punto de vista neurológico, se observa generalmente un retraso en la adquisición de los hitos del desarrollo y del lenguaje. A nivel cognitivo, la discapacidad suele ser leve-moderada, presentando durante la etapa escolar problemas de aprendizaje. Con la edad también se hacen evidentes las alteraciones del comportamiento propias de los pacientes con SPW, con tendencia a desarrollar comportamientos compulsivos (como pellizcarse la piel) o manipuladores, la presencia de rabietas frecuentes y trastornos por déficit de atención e hiperactividad, con comportamientos incluso agresivos, que se agravan conforme avanza la edad. Hasta en un 25% de los casos se han descrito comportamientos sugestivos de trastorno del espectro autista2. Los trastornos del sueño son también frecuentes, con aparición de apnea del sueño de origen central y obstructivo, con somnolencia diurna6. Otras de las características que podemos encontrar en estos individuos son un umbral para el dolor elevado, dificultad para vomitar, hipotiroidismo y alteración en la percepción y regulación de la temperatura2.
El diagnóstico del SPW se ha basado en una serie de criterios clínicos establecidos por consenso en 19937. En la actualidad, siendo el gold standard el diagnóstico genético, estos criterios clínicos se emplean para valorar cuáles son los individuos con riesgo de presentar SPW y susceptibles de realizar pruebas diagnósticas. En este sentido, Gunay7 llevó a cabo una revisión retrospectiva en pacientes con SPW confirmado mediante estudio genético para evaluar la validez y sensibilidad de los criterios diagnósticos establecidos hasta la fecha. Observó que, dentro de los criterios mayores, los que se presentaban con más frecuencia eran la hipotonía neonatal y el retraso global del desarrollo. En base a sus hallazgos establecieron una serie de criterios clínicos en función de la edad (Tabla 1), cuya presencia debe hacernos sospechar que nos encontramos ante un posible caso y llevar a cabo el estudio genético7. Para realizar este estudio se llevará a cabo el estudio del cariotipo y el test de metilación, que constituye la prueba inicial de cribado y nos permite confirmar el diagnóstico del síndrome, aunque no distinguir entre deleción, disomía uniparental o defecto de impronta3. Para ello será necesario aplicar las técnicas de hibridación fluorescente in situ (FISH) y análisis de microsatélites. La técnica de FISH detecta si existe deleción de la región 15q11-q13, donde se sitúan los genes responsables del SPW, como ocurrió en el caso de nuestro paciente. En caso de que el FISH sea negativo, se llevará a cabo el análisis de los microsatélites que permite diferenciar si ambos cromosomas son de origen biparental, o por el contrario son ambos de origen materno, en cuyo caso estaríamos ante una disomía uniparental materna. Si el origen fuera biparental se tratará, por defecto, de una alteración en la impronta genómica3,8. La importancia de un correcto diagnóstico reside en poder ofrecer a los progenitores consejo genético para una descendencia futura, ya que el riesgo de recurrencia varía en función de la alteración genética subyacente. Así, en las microdeleciones y en la disomía uniparental materna el riesgo de recurrencia es menor del 1%, mientras que cuando hay defectos en la impronta genómica este puede ascender hasta el 50%4,8.

El tratamiento de este síndrome precisa un abordaje multidisciplinar. Durante los primeros meses de vida prima el soporte nutricional y el inicio precoz de la Atención Temprana. Durante la infancia comienzan los problemas en relación con la falta de saciedad y la tendencia a la ganancia excesiva de peso, precisando un control muy estricto de la alimentación y el fomento de la actividad física para evitar las complicaciones de la obesidad5. Con respecto al tratamiento con hormona del crecimiento de estos pacientes, este ha demostrado causar una mejoría del crecimiento lineal y de la composición corporal, así como de la densidad ósea y perfil lipídico, aunque su uso debe individualizarse1. Su utilización se ha asociado, especialmente en pacientes con factores de riesgo (obesidad severa, apnea del sueño o infecciones respiratorias de repetición), con un aumento de la mortalidad durante las primeras semanas de tratamiento debido al empeoramiento de los problemas respiratorios, especialmente de la apnea del sueño, por lo que una adecuada evaluación respiratoria con pulsioximetría o polisomnografía nocturna es imprescindible antes de iniciar el mismo1,6.
La obesidad mórbida es la principal causa de morbimortalidad durante la edad adulta de los pacientes afectados de este síndrome, debido a complicaciones cardiovasculares y respiratorias2. Durante la infancia se ha descrito como principal causa de mortalidad los episodios de atragantamiento y aspiración, probablemente en relación con la ansiedad por la comida unida a cierta hipotonía e incoordinación oromotora6.
Conclusión
El conocimiento de las características fenotípicas y clínicas del SPW en el periodo neonatal es fundamental para poder establecer un diagnóstico de certeza precoz, que nos ayudará a la hora de realizar un correcto abordaje multidisciplinar y seguimiento de esta enfermedad, con el objetivo de intentar así mejorar a largo plazo el pronóstico de los pacientes afectados. El consejo genético, basado en los hallazgos encontrados en los estudios cromosómicos, es fundamental para poder realizar un pronóstico de recurrencia del cuadro en posteriores gestaciones.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS: FISH: hibridación fluorescente in situ · GH: hormona del crecimiento · IGF-1: factor de crecimiento similar a la insulina tipo 1 · SPW: síndrome de Prader-Willi.
BIBLIOGRAFÍA
- Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38:1249-63.
- Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17:3-13.
- Kalsner L, Chamberlain SJ. Prader-Willi, Angelman, and 15q11-q13 duplication syndromes. Pediatr Clin N Am. 2015;62:587-606.
- Moreno García M, Barreiro Miranda E. Impronta genómica. An Esp Pediatr. 1998;48:567-74.
- Scheimann AO. Clinical features, diagnosis, and treatment of Prader-Willi syndrome. En: UpToDate [en línea] [consultado el 31/05/2017]. Disponible en www.uptodate.com/contents/clinical-features-diagnosis-and-treatment-of-prader-willi-syndrome
- Miller J, Silverstein J, Shuster J, Driscoll DJ, Wagner M. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi syndrome. J Clin Endocrinol Metab. 2006;91:413-7.
- Gunay AM, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB. The changing purpose of Prader-Willi Syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108:e92.
- Scheimann AO. Epidemiology and genetics of Prader-Willi syndrome En: UpToDate [en línea] [consultado el 31/05/2017]. Disponible en www.uptodate.com/contents/epidemiology-and-genetics-of-prader-willi-syndrome
Comentarios
Este artículo aún no tiene comentarios.