
Vol. 13 - Num. 51
Revisiones
Genitales ambiguos
Francisco Pelayo Baezaa, Iván Carabaño Aguadob, Francisco José Sanz Santaeufemiac, Enrique La Orden Izquierdod
aServicio de Pediatría. Hospital Infanta Elena. Valdemoro. Madrid. España.
bSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
cSección de Pediatría Hospitalaria. Hospital Infantil Universitario Niño Jesús. Madrid. España.
dUnidad de Gastroenterología y Nutrición. Servicio de Pediatría. Hospital Infanta Elena. Valdemoro. Madrid. España.
Correspondencia: FJ Pelayo . Correo electrónico: franmarpb@msn.com
Cómo citar este artículo: Pelayo Baeza FJ, Carabaño Aguado I, Sanz Santaeufemia FJ, La Orden Izquierdo E. Genitales ambiguos. Rev Pediatr Aten Primaria. 2011;13:419-33.
Publicado en Internet: 11-10-2011 - Número de visitas: 80018
Resumen
El nacimiento de un niño con genitales ambiguos representa un choque emocional para la familia y un reto para los médicos, cuando no una verdadera situación de urgencia en etapa neonatal.
Los trastornos congénitos que dan lugar a una discrepancia entre genitales externos, gónadas y sexo cromosómico son clasificados como anomalías o trastornos de la diferenciación sexual (ADS). En el año 2006 se decidió consensuar la terminología a emplear, desechando aquellos términos que resultaban peyorativos, tales como intersexo, hermafroditismo o pseudohermafroditismo, etc. Desde entonces hablamos de ADS.
Presentan una incidencia anual de 1/4500 nacidos vivos, aproximadamente, siendo la hiperplasia suprarrenal congénita (HSC) la causa más frecuente. Esta incidencia aumenta al incluir los casos de criptorquidia, micropene e hipospadias neonatales.
La evaluación de estos niños debe realizarse de forma precoz, debido a que la forma más frecuente de estas alteraciones es la HSC, que puede poner en riesgo la vida del neonato.
La valoración inicial debe incluir exploración física, determinación de iones, 17-hidroxiprogesterona, cortisol, 11-desoxicortisol, 17-hidroxipregnenolona, dehidroepiandrosterona y hormona adrenocorticotropa para valorar la posibilidad de HSC. También debe solicitarse cariotipo, gen SRY y ecografía abdominal.
En función de los resultados se clasificarán estos trastornos en ADS 46XX, ADS 46XY y ADS por alteraciones cromosómicas.
El manejo de dicha situación debe ser realizado por un equipo multidisciplinario (neonatólogos, genetistas, endocrinólogos pediátricos, cirujanos pediátricos y psicólogos) en hospitales con experiencia.
Palabras clave
● Anomalías de la diferenciación sexual ● Genitales ambiguos ● GónadasDefinición de las anomalías de la diferenciación sexual
El conjunto de trastornos congénitos que dan lugar a una discrepancia entre genitales externos, gónadas y sexo cromosómico se denomina anomalías o trastornos de la diferenciación sexual (ADS)1.
Presentan una incidencia anual de aproximadamente 1/4500 nacidos vivos2. Se incluyen criptorquidia bilateral, hipospadias perineal con escroto bífido, clitoromegalia, fusión labial posterior, fenotipo femenino con gónada palpable (con hernia inguinal asociada o sin ella) e hipospadias con criptorquidia unilateral.
Clasificación de las anomalías de la diferenciación sexual (2006)
En el año 2006 se decidió consensuar la terminología a emplear, desechando aquellos términos que resultaban peyorativos, tales como intersexo, hermafroditismo/pseudohermafroditismo, etc. Desde entonces se habla de ADS1 (tabla 1).

Evaluación inicial
La presencia de genitales externos anómalos que impiden asignar sexo masculino o femenino obliga a la realización de una minuciosa historia clínica, con recogida exhaustiva de antecedentes familiares, exploración física detallada y pruebas complementarias.
Hasta que no tengamos resultados sobre cariotipo y 17-hidroxiprogesterona (17-OH-PG), mantendremos al neonato en observación, con realización diaria de gasometría e iones hasta descartar la hiperplasia suprarrenal congénita (HSC), patología que en su forma pierde-sal supone un riesgo vital para el neonato.
Recogida de antecedentes familiares
Preguntar sobre exposición prenatal a andrógenos, virilización materna durante el embarazo, insensibilidad androgénica familiar (mujeres con amenorrea o pubertad retrasada), muertes neonatales inexplicadas (HSC) e historia de consanguinidad.
Exploración física
La exploración física de los genitales externos (tamaño del clítoris o del pene, separación de los labios mayores, grado de fusión del rafe medio, bolsas escrotales, situación del meato urinario, abocamiento vaginal), la palpación de las gónadas (en bolsas, labios mayores o conducto inguinal) y su posible nivel de descenso permiten establecer el grado de ambigüedad de acuerdo con los estadios descritos por Prader4.
- Longitud y grosor del pene: la longitud debe medirse por su cara dorsal, desde el pubis (comprimiendo con la regla la grasa suprapúbica) al extremo distal del pene, excluyendo el prepucio. El grosor se medirá en su tercio medio. En el recién nacido a término, la longitud normal es mayor o igual a 2,5 cm y su diámetro mayor o igual a 0,9 cm. Otras causas de micropene, además de déficit de testosterona en el segundo o tercer trimestre de gestación, son el déficit de hormona de crecimiento (GH) o de gonadotropina coriónica humana (HCG). El micropene con hipospadias es frecuentemente secundario a deficiencia de GH o HCG.
- Clítoris: la anchura del clítoris se mide presionando suave, pero firmemente, con el dedo pulgar e índice para eliminar el exceso de piel y tejido subcutáneo. La medida normal en neonatos a término es de 2 a 6 mm. La longitud del mismo varía dependiendo del grupo étnico, pero si es mayor de 0,9 cm tendremos que sospechar una posible ambigüedad genital o virilización. En recién nacidos pretérmino, el clítoris puede aparentar ser más prominente porque el tamaño del mismo está totalmente desarrollado a las 27 semanas de gestación y existe menos grasa en los labios mayores. La clitoromegalia secundaria a exposición a andrógenos en sujeto 46XX puede ser causada por HSC, ADS testicular, exposición materna a los andrógenos o, raramente, por infiltraciones tumorales en el clítoris, como en la neurofibromatosis. Algunos autores proponen el índice longitud por anchura (mm) para valorar la exposición a los andrógenos, aunque como regla general este cálculo no es necesario para la valoración clínica.
- Gónadas: el escroto, los labios mayores y el área inguinal deben ser cuidadosamente explorados para identificar la presencia y posición de las gónadas. En un sujeto 46XY la ausencia de testículos en la bolsa escrotal puede deberse al síndrome de persistencia del conducto mülleriano o anorquia. En un sujeto 46XX habría que descartar como primera opción la HSC.
- Apertura uretral: un orificio uretral en la base del pene puede ser orientativo y debemos descartar desde hipospadias simple a un seno urogenital virilizado. Estos hallazgos deben ser confirmados con pruebas de imagen como cistografía o vaginoscopia, porque el examen físico puede ser engañoso.
- Ratio anogenital: es la distancia entre el ano y la comisura posterior de los labios menores dividida entre la distancia entre el ano y la base del clítoris/falo. Una ratio > 0,5 sugiere virilización.
- Datos clínicos, además de la ambigüedad genital franca, que sugieren trastornos de la diferenciación sexual3 son: a) criptorquidia bilateral; b) micropene (< 2,5 cm en recién nacidos a término), el micropene sin hipospadias asociado no se considera genital ambiguo, pero puede reflejar otros trastornos; c) hipospadias perineal con escroto bífido; d) hipospadias y gónada unilateral no palpable; e) clitoromegalia (ancho > 6 mm y/o longitud > 9 mm); f) fusión labial posterior (ratio anogenital > 0,5), y g) gónadas palpables en pliegues labioescrotales.
Exploraciones complementarias
- Cariotipo y gen SRY: la presencia de genitales ambiguos obliga a la realización de un cariotipo de sangre periférica de forma urgente (resultados en 3-4 días). El cariotipo es realizado a partir de leucocitos periféricos. Siempre es aconsejable el estudio de 200 células por lo menos, debido a la posibilidad de mosaicismos. También debe determinarse la existencia del gen SRY, pues su existencia en individuos 46XX (translocación) produce virilización y su ausencia en 46XY feminización (delección).
- A partir de las 48 horas de vida (nunca antes) deberá realizarse una analítica solicitando:
- Gasometría venosa, glucosa, ionograma.
- Hormona adrenocorticotropa (ACTH), cortisol y estudio de esteroides suprarrenales y gonadales:
- 17-OH-PG: elevada en la HSC, siendo el déficit de 21-hidroxilasa la causa más frecuente de ambigüedad genital en 46XX; además, representa una urgencia vital en su forma de síndrome pierde-sal. Otras causas de HSC, menos frecuentes, son el déficit de 11-betahidroxilasa y de 3-beta hidroxiesteroide deshidrogenasa (que es causa de genitales ambiguos en 46XX y XY). Para el estudio de estas formas menos comunes de HSC solicitaremos: dehidroepiandrosterona (DHEA), androstendiona, progesterona, 17-hidroxipregnenolona, 11-desoxicortisol y desoxicorticosterona.
- Testosterona y dihidrotestosterona (DHT): para el diagnóstico de los ADS 46XY.
- Gonadotropinas basales (hormona foliculoestimulante, hormona luteinizante [LH]): en el hipogonadismo hipogonadotropo los valores son indetectables. Valores por encima del percentil 95 orientan a un fallo gonadal primario. En mutaciones del receptor androgénico y en la hipoplasia de las células de Leidig nos encontraremos con valores elevados de LH.
- Hormona antimülleriana (HAM): valora la presencia de tejido testicular.
- Ecografía abdominopélvica: mientras llegan los resultados, para intentar identificar restos müllerianos, útero, ovarios y testículos abdominales o en el trayecto inguinal.
- Resonancia abdominopélvica: es más precisa, pero requiere gran experiencia y sedar al neonato.
Algoritmo diagnóstico
En función del resultado del cariotipo, las ADS se clasifican en uno de los tres grupos siguientes:
- Anomalías o trastornos de la diferenciación sexual en individuos 46XX.
- Anomalías o trastornos de la diferenciación sexual en individuos 46XY.
- Anomalías o trastornos de la diferenciación sexual por alteración de los cromosomas.
Anomalías o trastornos de la diferenciación sexual en individuos 46XX
Si el cariotipo es 46XX y los niveles de 17-OH-PG son elevados, deberemos descartar hiperandrogenismo por HSC. Niveles normales dirigirán las investigaciones hacia un exceso de andrógenos de origen materno o placentario. Descartado todo lo anterior, será obligada la laparoscopia con biopsia gonadal para buscar anomalías del desarrollo gonadal (figura 1).
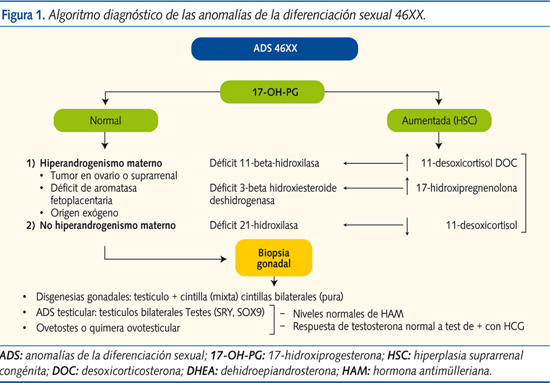
ADS 46XX por exceso de andrógenos
- Origen fetal:
- La HSC es la causa más común de virilización en 46XX, siendo el déficit de 21-hidroxilasa la causa más frecuente (90-95%). Se objetiva un aumento de 17-OH-PG, ACTH y andrógenos suprarrenales, con la subsecuente virilización. En un 70% de los casos nos encontramos ante una forma pierde-sal, con alteraciones hidroelectrolíticas (hiponatremia, hiperpotasemia, deshidratación) y disminución del cortisol plasmático.
- Existen otros déficits enzimáticos que cursan con elevación de la 17-OH-PG y genitales ambiguos en individuos XX. Por tal motivo, para conocer a qué nivel de la esteroidogénesis se ha producido el fallo, es imprescindible conocer los niveles de 17-hidroxipregnenolona, 11-desoxicortisol, desoxicorticosterona y DHEA.
- El déficit de 11-betahidroxilasa (segunda causa más frecuente de HSC) cursa con aumento del 11-desoxicortisol y la desoxicorticosterona. Se producen genitales ambiguos, elevación de la tensión arterial por aumento de la desoxicorticosterona con actividad mineralocorticoide (por este motivo no hay un síndrome pierde-sal) y supresión de la renina.
- El déficit de 3-beta hidroxiesteroide deshidrogenasa (< 1-2% de todas las HSC) cursa con aumento de la 17-hidroxipregnenolona y la DHEA (produce ambigüedad genital en sujetos 46XX y XY y síndrome pierde-sal).
- Origen fetoplacentario, por déficit de aromatasa y de óxido-reductasa.
- Origen materno, en cuyo caso hay que descartar la presencia de tumores maternos virilizantes (luteoma, quistes teca-luteínicos, tumores suprarrenales, etc.), HSC incorrectamente tratada y toma de fármacos androgénicos.
ADS 46XX por anomalías del desarrollo gonadal
Si los estudios previos son negativos, deberán realizarse una laparoscopia y con biopsia gonadal para llegar al diagnóstico de confirmación de disgenesia gonadal, 46XX idiopático (tienen ovarios), ovotestes o ADS testicular. Causas de ovotestes en 46XX son: a) presencia de gen SRY o duplicación del gen SOX9; b) mutación de genes autosómicos o del cromosoma X, que permite la determinación testicular incompleta aun en ausencia del gen SRY, o c) existencia de una línea celular ignorada que lleve todo o parte del cromosoma Y.
Anomalías o trastornos de la diferenciación sexual en individuos 46XY8
El diagnóstico de ADS en un individuo 46XY es más complicado debido a la gran variabilidad fenotípica y al número extenso de causas que pueden originarlas. Es importante recordar que algunos tipos de HSC poco habituales pueden producir genitales ambiguos en sujetos 46XY, tales como el déficit de 17-alfa-hidroxilasa/17-20-liasa (solo genitales ambiguos en XY) y 3-beta hidroxiesteroide deshidrogenasa (genitales ambiguos en XX y XY), además de las mutaciones de P450-óxido reductasa y déficit proteína StaR (hiperplasia lipoidea).
Una vez conozcamos que el cariotipo es 46XY, procederemos a objetivar si existe alteración en la biosíntesis de testosterona (respuesta de testosterona tras test de estímulo con gonadotropina coriónica [beta-HCG]). Cuando se objetiva un defecto de la síntesis de testosterona, también debe ser evaluada la función suprarrenal (test de estímulo con ACTH) para localizar el lugar del defecto enzimático (suprarrenal o testicular)3. La HAM, que es un marcador de la existencia de tejido testicular, nos ayudará al diagnóstico (figura 2).
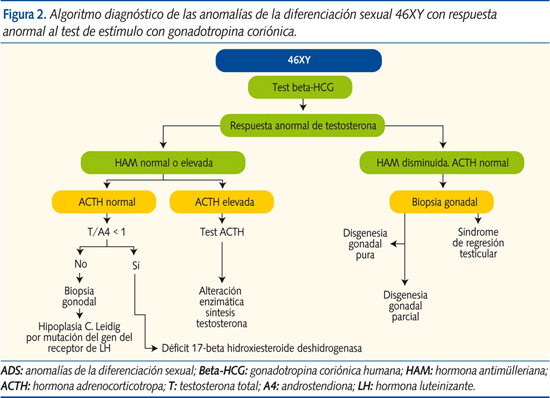
Anomalías de la diferenciación sexual 46XY con respuesta anormal de testosterona al test de estímulo con beta-HCG
- Sin defecto enzimático suprarrenal (ACTH normal):
- Con HAM en niveles normales:
- Hipoplasia/aplasia de las células de Leydig por mutación del gen LHCGR: presenta niveles elevados de LH y disminución de la síntesis de testosterona con defecto en la virilización. Cursa con disminución de la síntesis de testosterona (test de estimulación con HCG con valores anormalmente bajos de testosterona; en la aplasia de células de Leydig, niveles ausentes) y niveles normales o aumentados de la HAM. Una ACTH basal y un test de ACTH en valores normales descartan alteración enzimática suprarrenal. La relación testosterona/androstendiona > 1 orienta al diagnóstico, que se confirmará con biopsia gonadal y estudio molecular del gen.
- Alteración enzimática testicular de las síntesis de testosterona: la alteración de la enzima 17-beta hidroxiesteroide deshidrogenasa produce niveles de testosterona disminuidos y androstendiona elevada (precursor previo al bloqueo enzimático). La ratio testosterona/androstendiona es generalmente < 0,8-1 tras estímulo con HCG. La HAM se encuentra normal o elevada y la ACTH permanece en niveles normales con test de ACTH normal por no tratarse de un fallo suprarrenal. Requiere confirmación con estudio molecular del gen.
- Con HAM disminuida: disgenesia gonadal pura, parcial y síndrome de regresión testicular: cursan con valores nulos/disminuidos de testosterona, tanto basales como tras estímulo con beta-HCG, y con HAM prácticamente indetectable. Estudio de función suprarrenal normal (ACTH basal y test de estímulo de ACTH normales). Se diagnostica mediante biopsia, es importante el estudio del gen SRY.
- Con HAM en niveles normales:
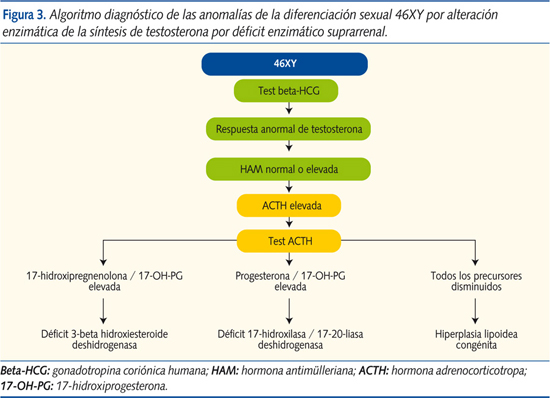
- Con defecto enzimático suprarrenal (ACTH elevada y test de ACTH alterado):
- Con HAM en niveles normales:
- Déficit de 3-beta hidroxiesteroide deshidrogenasa: produce genitales ambiguos en sujetos XX (acumulación de DHEA) y XY (defecto de virilización por fallo en la conversión de la DHEA en androstendiona, precursor de la testosterona), además de síndrome pierde-sal (aumenta la renina y disminuye la aldosterona). Este defecto se distingue por aumento de la ratio 17-hidroxi-pregnenolona/17-OH-PG y del cociente DHEA/androstendiona, tras estímulo con ACTH. Cursa con HAM intacta.
- Déficit a nivel de la enzima 17-hidroxilasa/17-20-liasa: produce falta de virilización en el varón, junto con hipertensión arterial e hiperpotasemia. Tras estímulo con ACTH se objetiva aumento de la relación progesterona/17-OH-PG con HAM en niveles normales.
- Hiperplasia lipoidea congénita: genitales ambiguos o fenotipo externo femenino en 46XY. Cursa con aumento destacado de ACTH con déficit generalizado de todos los precursores suprarrenales, también tras estímulo de ACTH, con disminución de la producción de andrógenos, e insuficiencia suprarrenal con disminución del cortisol y síndrome pierde-sal. HAM inalterada (figura 3).
- Con HAM en niveles normales:
Anomalías de la diferenciación sexual 46XY con respuesta normal de testosterona al test de estímulo con gonadotropina humana (beta-HCG) (figura 4)
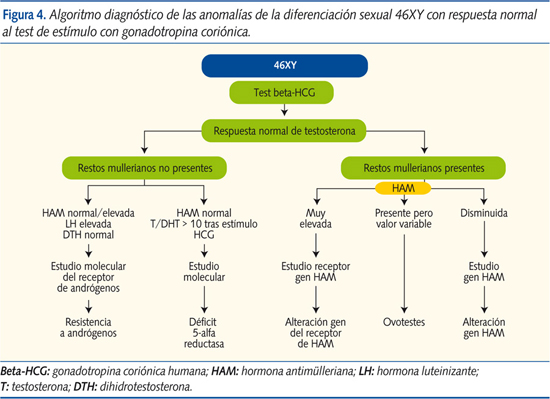
- Con restos müllerianos ausentes:
- Resistencia a andrógenos (causa más frecuente de ADS 46XY). El feto 46XY presenta falta de masculinización por alteración del receptor de los andrógenos en los tejidos periféricos. Presenta gónadas masculinas con ausencia de restos müllerianos y HAM en niveles normales/levados. La LH, la testosterona basal y tras estímulo de HCG, así como la DHT, están en niveles normales. Requiere estudio molecular del gen del receptor androgénico para llegar al diagnóstico.
- Déficit de 5-alfa-reductasa: enzima cuyo déficit impide la formación de DHT a partir de la testosterona, con la consiguiente falta de virilización. Presenta niveles de testosterona basales y tras estímulo con HCG normales, con HAM presente. El cociente testosterona/DHT es > 10 tras estímulo con HCG. No es evidenciable ningún resto mülleriano y tienen gónadas masculinas bilaterales intactas con fenotipo femenino. HAM normal. El estudio molecular confirma el diagnóstico.
- Con restos müllerianos presentes:
- Alteración del gen de la hormona antimülleriana y del gen del receptor de la misma: la HAM es producida en las células de Sertoli del testículo, inhibiendo el desarrollo de los conductos de Müller. En ocasiones, su síntesis o su receptor se encuentran alterados, situación que conlleva el desarrollo de las estructuras müllerianas y ambigüedad genital del 46XY con gónadas bilaterales masculinas (generalmente criptorquia) y/o hernia uteroinguinal. Presentan testosterona basal y tras estímulo normal. La existencia de una HAM anormalmente elevada (alteración del gen del receptor) o muy disminuida (alteración del gen de la HAM), además del estudio molecular del gen afectado, nos llevan al diagnóstico.
- Ovotestes: presencia de tejido ovárico y testicular con testosterona en niveles masculinos.
Anomalías o trastornos de la diferenciación sexual en individuos con alteraciones cromosómicas3
Incluyen 45X y mosaicos (síndrome de Turner y variantes), 47XXY (síndrome de Klinefelter y variantes), 45X/46XY (disgenesia gonadal mixta o disgenesia gonadal asimétrica) y 46XX/46XY (ADS ovotesticular, quimerismo).
Este subtipo de ADS también incluye alteraciones que no presentan ambigüedad sexual, como el síndrome de Turner 45X0 y el síndrome de Klinefelter 47XXY, y alteraciones que pueden presentar genitales ambiguos, como individuos con mosaico 45X/46XY, 46XX/46XY, etc.
- Disgenesia gonadal mixta (45X0/46XY): puede presentar diferentes grados de ambigüedad genital con cariotipo también variable. Se caracteriza por presentar una anatomía del aparato reproductor asimétrica. Es frecuente objetivar en un lado un desarrollo testicular con estructuras wolfianas escasamente desarrolladas y en el otro lado una cintilla gonadal con estructuras müllerianas rudimentarias.
- El riesgo de gonadoblastoma se encuentra incrementado, sobre todo en los casos en que la gónada se encuentra en localización intraabdominal, donde debe plantearse la gonadectomía. Si la gónada se encuentra en el escroto, deberá realizarse un seguimiento estrecho con ecografía y biopsia gonadal para detectar la transformación tumoral.
- ADS ovotesticular (46XX/46XY), antiguamente denominado hermafroditismo verdadero. Se caracteriza por la presencia de testículo y ovario o gónadas con ambos tipos de tejidos (ovotestes). Asimismo, es común la existencia de estructuras procedentes tanto del conducto de Müller como de Wolfram. Más frecuentemente asociado con cariotipo XX, pero puede verse también en cariotipo XY y mosaicismos (XX/XY). El cariotipo y laparoscopia con biopsia gonadal nos dan el diagnóstico.
Tratamiento4
El tratamiento de los genitales ambiguos debe ser realizado por un equipo multidisciplinario en hospitales con experiencia (neonatólogo, endocrino pediátrico, cirujano pediátrico y psicólogo). Ante un recién nacido 46XX con genitales ambiguos, la correcta adjudicación del sexo civil femenino es rápida, sobre todo si se trata de una HSC. El resto de casos puede ser mucho más complicado. La reconstrucción hacia el sexo femenino, en general, es más sencilla desde el punto de vista quirúrgico; además, la posibilidad de malignización de las gónadas disgenéticas aconseja su extirpación.
El tratamiento quirúrgico se deberá realizar en función del sexo civil asignado, con feminización de los genitales externos y extirpación de las gónadas que no sean ovarios, en el caso de que la elección haya sido sexo femenino.
En caso de elección de sexo masculino, la reconstrucción precisa un tamaño mínimo de los cuerpos cavernosos, corrección del hipospadias y descenso a bolsa o conducto inguinal de las gónadas masculinas (o su extirpación en caso de que no sea posible conservarlas). En ausencia de gónadas será necesario colocar sendas prótesis testiculares con fines estéticos.
En cuanto al tratamiento hormonal, los pacientes de ambos sexos deberán recibir tratamiento hormonal sustitutivo para el desarrollo de los genitales sexuales secundarios, en la edad de inducción de la pubertad (11,5-12 años de edad ósea en las niñas y 12,5-13 años de edad ósea en los niños). Dicho tratamiento deberá continuarse en la edad adulta.
Cuando en edades tardías el sexo civil asignado es incorrecto, desde el punto de vista quirúrgico y funcional, su cambio es muy difícil, creando una situación psicológica muy negativa por la cronología conocida de la adquisición del sexo mental.
Bibliografía
- Lee PA, Houk CP, Almed SF. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118:e488.
- Hughes IA, Nihoul-Fékété C, Thomas B, Cohen-Kettenis PT. Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2007;21:351-65.
- Houk CP, Levitsky LL. Evaluation of the infant with ambiguous genitalia. Uptodate [consultado el 25/06/2011]. Disponible en www.uptodate.com
- Pombo M. Tratado de Endocrinología Pediátrica. Madrid: McGraw Hill; 2009. p. 583-608.
- Ahmed SF, Hughes IA. The genetics of male undermasculinization. Clin Endocrinol (Oxf). 2002;56:1-18.
- Clarkson MJ, Harley VR. Sex with two SOX on: SRY and SOX9 in testis development. Trends Endocrinol Metab. 2002;13:106-11.
- Achermann JC, Meeks JJ, Jameson JL. Phenotypic spectrum of mutations in DAX-1 and SF-1. Moll Cell Endocrinol. 2001;185:17-25.
- Argente Oliver J, Soriano Guillén L. Manual de Endocrinología pediátrica. Madrid: Ergon; 2010. p. 97-107.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflicto de intereses en relación con la preparación y publicación de este artículo.