
Vol. 27 - Num. 107
Notas clínicas
Síndrome de Gitelman: diagnóstico a partir de un hallazgo casual de hipopotasemia
M.ª Tatiana Fernández Garridoa, Anny Vanessa Martínez Báezb, Jesús M.ª Pascual Pérezc, Jorge Lévano Vásquezc, M.ª Mercedes Santos Herreroa
aMédico de Familia y Médico Puericultor. CS Los Pintores. Parla. Madrid. España
bMédico de Familia. CS Los Pintores. Parla. Madrid. España
cPediatra. CS Los Pintores. Parla. Madrid. España.
Correspondencia: MT Fernández. Correo electrónico: tatiana.fernandez@salud.madrid.org
Cómo citar este artículo: Fernández Garrido MT, Martínez Báez AV, Pascual Pérez JM, Lévano Vásquez J, Santos Herrero MM. Síndrome de Gitelman: diagnóstico a partir de un hallazgo casual de hipopotasemia . Rev Pediatr Aten Primaria. 2025;27:263-7. https://doi.org/10.60147/907ee68e
Publicado en Internet: 15-07-2025 - Número de visitas: 8021
Resumen
El síndrome de Gitelman es una tubulopatía de herencia autosómica recesiva debida a mutaciones en el gen SLC12A3, encargado de codificar transportadores de NaCl y Mg en la membrana apical de las células del túbulo contorneado distal.
Se caracteriza por cursar con hipopotasemia, alcalosis metabólica, hipocalciuria, hipomagnesemia e hiperaldosteronismo hiperreninémico.
El diagnóstico se basa en los síntomas clínicos y en las alteraciones analíticas previamente descritas. Los síntomas pueden variar, desde leves (calambres, debilidad) o incluso pacientes asintomáticos, hasta síntomas graves (tetania, convulsiones). La confirmación diagnóstica se realiza mediante estudio genético.
El diagnóstico diferencial incluye el síndrome de Bartter tipo III y la hipomagnesemia renal con hipocalciuria.
El tratamiento es sintomático, mediante suplementación con potasio y magnesio.
Palabras clave
● Hipopotasemia ● Síndrome de Bartter ● Síndrome de GitelmanINTRODUCCIÓN
El síndrome de Gitelman (SG) forma parte de las tubulopatías perdedoras de sal. Se caracteriza por hipopotasemia, hipomagnesemia, alcalosis metabólica, normocalcemia, hipocalciuria y presión arterial normal o ligeramente baja con niveles elevados de renina y aldosterona1,2.
Se estima una prevalencia de 1 por 40 000 individuos, lo que lo convierte en uno de los desórdenes tubulares renales hereditarios más frecuentes. Presenta un patrón de herencia autosómica recesiva.
La presentación clínica es muy variable, abarcando desde pacientes con manifestaciones muy evidentes, hasta formas leves o asintomáticas. La edad de inicio también es muy heterogénea, suele ser en la adolescencia o edad adulta, aunque se ha descrito también en la infancia e incluso en el periodo neonatal3.
Aunque se considera una entidad de curso generalmente benigno, la combinación de hipopotasemia con hipomagnesemia puede prolongar el intervalo QT y desencadenar arritmias potencialmente graves que pueden amenazar la vida del paciente3.
El diagnóstico diferencial incluye el síndrome de Bartter tipo III y la hipomagnesemia renal con hipocalciuria. Por ello, establecer un diagnóstico preciso y confirmarlo mediante estudio genético resulta crucial tanto para el seguimiento del paciente como para el asesoramiento genético familiar4.
El tratamiento se basa en la administración de cloruro potásico y sales de magnesio. Se pueden asociar diuréticos ahorradores de potasio o antagonistas de la aldosterona en casos seleccionados5.
A continuación, se presenta el caso clínico de un paciente adolescente diagnosticado de SG a partir de un hallazgo casual de hipopotasemia en una analítica realizada por baja talla y bajo peso.
CASO CLÍNICO
Paciente varón de 14 años que acude a la consulta de Pediatría de Atención Primaria para la revisión del niño sano.
Durante la valoración, se objetivó talla baja y bajo peso para su edad y potencial genético.
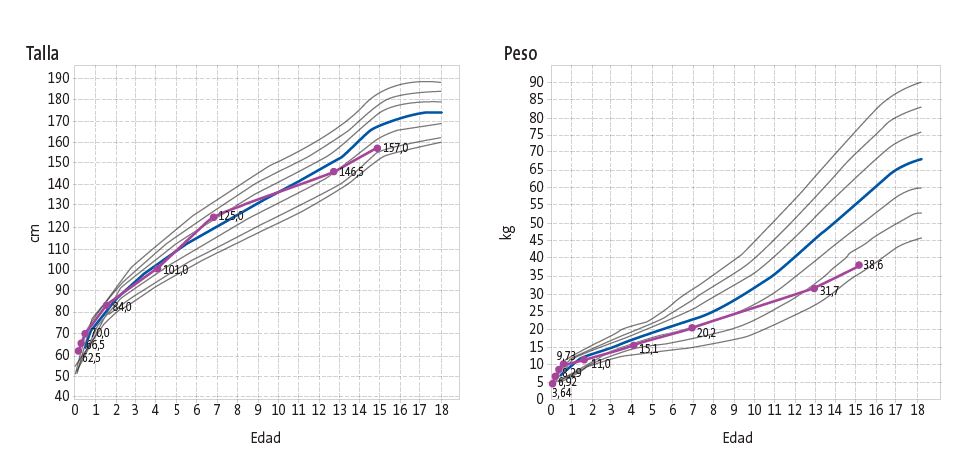
Los datos de la exploración física fueron: peso de 38,6 kg (p 3; -1,91 DE) y una talla de 157 cm (p 6; -1,63 DE), resultando en un IMC de 15,66 kg/m² (p 6; -1,61 DE y una superficie corporal de 1,3 m², lo que corresponde a un bajo peso severo. En cuanto al desarrollo puberal, el paciente se encontraba en estadio Tanner II.
La talla proyectada del paciente era de 167 cm, significativamente inferior a su talla diana de 188,5 ± 5 cm (p 95; +1,73 DE), calculada a partir de la talla paterna (188 cm; p 95; +1,65 DE) y materna (176 cm; p 97; +1,99 DE).
Al analizar su curva de crecimiento, se observó una disminución progresiva del percentil de talla, situándose entre los percentiles 50 y 75 hasta los 7 años para después descender hasta el percentil 10 en el momento de la revisión. Respecto al peso, pasó del percentil 90 en el primer año de vida, al percentil 25 a los 4 años y situándose finalmente en el percentil 10 en el momento de la revisión (Figura 1).
| Figura 1. Paciente de 14 años con síndrome de Gitelman: datos antropométricos |
|---|
 |

Tras la valoración en la consulta de Pediatría, se solicitó una analítica completa que incluyó hormonas tiroideas y estudio de celiaquía. De los resultados obtenidos, destacó una hipopotasemia severa, con un valor de potasio de 2,3 mmol/L (valores de referencia [VR]: 3,5-5,3 mmol/L), con normalidad del resto de parámetros. Ante este hallazgo, se decidió derivar al paciente al Servicio de Urgencias para confirmación analítica y valoración clínica.
A su llegada a urgencias, el paciente persistía asintomático (negaba debilidad muscular, mialgias, vómitos, diarrea, poliuria, coluria, palpitaciones o mareo) y no refería toma de fármacos, ni de forma crónica ni reciente. La exploración física fue rigurosamente normal.
Se realizó una nueva analítica, que confirmó la hipopotasemia severa, con un potasio de 2,2 mmol/L, manteniéndose el resto de iones dentro del rango normal. El electrocardiograma fue normal, presentando ritmo sinusal a 100 lpm, eje QRS a 80°, intervalos eléctricos normales (PR: 140 ms, QTc: 440 ms), repolarización conservada con T negativas en V1, sin signos de crecimiento de cavidades ni bloqueos. Alteración inespecífica de la repolarización, con leve descenso del ST en cara inferior. No presentaba datos de bloqueos ni de hipertrofia de cavidades.
Ante la persistencia de la hipopotasemia, se inició tratamiento con suplemento oral de potasio y se derivó a consultas externas de Nefrología para completar el estudio. Durante las revisiones en Nefrología se solicitaron nuevas pruebas complementarias.
La analítica sanguínea confirmó la persistencia de la hipopotasemia, con un potasio de 2,8 mmol/L. Se asoció además hipocloremia, con un cloruro de 95 mmol/L (VR: 99-109 mmol/L) e hipomagnesemia, con un magnesio de 1,58 mg/dL (VR: 1,6-2,4 mg/dL). La determinación de concentración de renina plasmática (CRP) en muestra obtenida en posición ortostática arrojó un valor de >500 mUI/mL (VR: 4,40-46,10 mUI/mL). La gasometría venosa reveló alcalosis metabólica compensada: pH 7,44 (VR: 7,33-7,42), pCO2: 55 mmHg (VR: 38-48 mmHg), HCO3-: 36,5 mmol/L (VR: 23-28 mmol/L), pO2: 26 mmHg (VR: 20-45 mmHg).
En el análisis de orina se detectó hipocalciuria, con un calcio urinario de 0,8 mg/dL (VR: 6,8-21,3 mg/dL).
La ecografía renal no evidenció nefrocalcinosis.
Finalmente, el estudio genético identificó dos variantes patogénicas en el gen SLC12A3, lo que confirmó el diagnóstico de SG.
El paciente continúa en seguimiento en el Servicio de Nefrología, con controles analíticos y clínicos periódicos, y mantiene suplementación oral de potasio.
DISCUSIÓN
El SG, descrito por primera vez en 1966, es una tubulopatía con un patrón de herencia autosómica recesiva, cuya prevalencia se estima en aproximadamente 1:40 0006.
Se caracteriza por la combinación de alcalosis metabólica, hipopotasemia, hipomagnesemia e hipocalciuria. Clínicamente, cursa con un estado de hipovolemia con hiperreninemia e hiperaldosteronismo, manteniendo una presión arterial normal o baja7.
La etiopatogenia del SG radica en mutaciones inactivantes del gen SLC12A3, que codifica el cotransportador NaCl sensible a tiazidas del túbulo contorneado distal. Existe una pérdida urinaria de ClNa con la subsiguiente depleción moderada de volumen y estimulación del sistema renina-angiotensina-aldosterona. Se evidencia una reducción en la expresión del canal de Mg2+ TRPM6 en la membrana apical de las células del túbulo contorneado distal que es responsable de la hipomagnesemia y un aumento en la reabsorción de Ca2+ que causa la hipocalciuria8.
La presentación clínica del SG es muy heterogénea, desde pacientes pediátricos con manifestaciones muy claras hasta adultos con síntomas muy leves, incluso casos asintomáticos cuyo diagnóstico se realiza a través de estudio genético9.
Los síntomas más frecuentes, derivados de las alteraciones electrolíticas características del síndrome, incluyen: debilidad muscular, calambres, fatiga y, en casos graves, arritmia cardíaca. La depleción de potasio y magnesio prolonga la duración del potencial de acción en los cardiomiocitos, lo que resulta en un intervalo QT prolongado en aproximadamente el 50% de los pacientes, lo que podría conducir a un mayor riesgo de arritmias ventriculares2. Espasmos musculares, temblores y convulsiones, en relación con el déficit de magnesio. Otros síntomas incluyen hipoventilación y confusión por alcalosis metabólica, así como aumento de la apetencia por alimentos salados debido a la pérdida renal de sodio.
También se puede observar retraso ponderoestatural y puberal, posiblemente asociado al desequilibrio electrolítico crónico, aunque el mecanismo exacto aún se desconoce. La ganancia de talla y peso mejora con la suplementación de los electrolitos deficitarios10.
Incluso podemos encontrarnos pacientes asintomáticos y son los hallazgos analíticos los que nos llevan al estudio y al diagnóstico9.
Entre las posibles complicaciones se incluyen condrocalcinosis y calcificaciones esclerocoroideas.
El diagnóstico del SG se basa en los síntomas clínicos y las alteraciones analíticas características en sujetos normotensos y en ausencia de ingesta de diuréticos, con confirmación mediante pruebas genéticas. En ocasiones, el diagnóstico puede ser incidental, tras una analítica sanguínea realizada por otros motivos.
En el diagnóstico diferencial se incluyen el síndrome de Bartter tipo III y la hipomagnesemia renal con hipocalciuria.
Los síndromes de Bartter y Gitelman son tubulopatías hereditarias estrechamente relacionadas. El SG tiene un fenotipo más leve y una presentación más tardía que el de Bartter, pudiendo diagnosticarse en la edad adulta. Ambos síndromes se caracterizan por hipopotasemia y alcalosis metabólica; el SG se distingue por presentar hipomagnesemia e hipocalciuria, mientras que en el de Bartter la excreción de calcio urinario suele estar normal o incluso aumentada11.
Respecto al diagnóstico diferencial con la hipomagnesemia renal con hipocalciuria, esta se diferencia del SG en que la concentración de potasio se mantiene en valores dentro del rango de referencia.
El tratamiento del SG se basa en la administración oral de cloruro potásico y sales de magnesio para corregir los desequilibrios electrolíticos.
El asesoramiento genético es fundamental. Dado su patrón de herencia autosómica recesiva, el riesgo de recurrencia en futuros embarazos es del 25%. En el caso de que los padres tengan otros hijos que no están clínicamente afectados, no se puede descartar que presenten SG, ya que las manifestaciones pueden aparecer más tarde. Es posible el diagnóstico prenatal mediante biopsia corial o líquido amniótico, para descartar dicha alteración en el feto12.
El pronóstico a largo plazo es excelente. Hasta la fecha, no hay evidencia de que el SG afecte a la esperanza de vida de los pacientes.
CONCLUSIÓN
Se puede concluir que el SG se trata de una enfermedad rara. Dada la baja frecuencia de esta enfermedad y a lo inespecífico de su presentación clínica, donde los pacientes pueden estar asintomáticos durante largos períodos de tiempo o presentar síntomas leves, es frecuente que existan retrasos en el diagnóstico.
La capacidad de reconocer las alteraciones electrolíticas clave, como la hipopotasemia persistente o la hipomagnesemia con la hipocalciuria, es fundamental. Estas anomalías, detectadas en la analítica sanguínea y de orina, deben guiar la sospecha diagnóstica hacia una tubulopatía.
La derivación oportuna a la consulta especializada de Nefrología para un estudio más profundo, incluyendo la confirmación genética, garantiza un diagnóstico preciso, un inicio temprano del tratamiento y la prevención de posibles complicaciones graves de esta enfermedad. Con una adecuada reposición electrolítica, es posible que los afectados alcancen valores iónicos cercanos a la normalidad y mejoren sus síntomas.
Es fundamental que el pediatra de Atención Primaria mantenga una alta vigilancia ante anomalías en las curvas de crecimiento (retraso ponderoestatural) y considere siempre la posibilidad de una enfermedad orgánica no endocrinológica en niños con fallo de medro o retraso puberal. Concretamente, se debe pensar en una tubulopatía cuando se presenten episodios de debilidad, espasmos y/o alteraciones electrolíticas.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Todos los autores han contribuido de forma equivalente en la elaboración del manuscrito publicado.
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
CRP: concentración de renina plasmática · SG: síndrome de Gitelman · VR: valores de referencia.
BIBLIOGRAFÍA
- Lozano E, Merino JL, Espejo B, Paraíso V. Síndrome de Gitelman y gestación a término. NefroPlus. 2016;8(2):95-186.
- Konrad, M. (2025). Síndromes de Bartter y Gitelman en niños: manifestaciones clínicas, diagnóstico y tratamiento. UpToDate [en línea] [consultado el 05/03/2025]. Disponible en www.uptodate.com/contents/bartter-and-gitelman-syndromes-in-children-clinical-manifestations-diagnosis-and-management
- Andrés MJ, Ansón M, Puente JJ. Síndrome de Gitelman en un paciente pediátrico: a propósito de un caso. Rev Lab Clin. 2017;10(4):208-211. https://doi.org/10.1016/j.labcli.2017.06.001
- Blanchard A, Bochkenhauer D, Bolignano D, Caló l, Cosyns É, Devusyst O, et al. Gitelman síndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversis Conference. Kidney Int. 2017;91(1):24-33. https://doi.org/10.1016/j.kint.2016.09.046
- García E, Jiménez M, De la Cerda F. Retraso de crecimiento y desarrollo puberal en el síndrome de Gitelman. Med Clin. 2021;157(12):588-9. https://doi.org/10.1016/j.medcli.2021.01.025
- Lozano E, Merino JL, Espejo B, Paraíso V. Síndrome de Gitelman y gestación a término. NefroPlus 2016;8(2):166-9.
- Márquez N, Villamil LP, Restrepo C. Síndrome de Gitelman: Importancia de la historia clínica para su diagnóstico. Reporte de caso. Salutem Sci Spiritus. 2015;1(2):38-43.
- Luis MI, García PM, García V. Tubulopatías. Sociedad Española de Nefrología. En: Nefrología al Día [en línea] [consultado 05/03/2025]. Disponible en www.nefrologiaaldia.org/es-articulo-tubulopatias-253
- Martín V, Lafarga MA, García l, Rodrigo MD. Diagnóstico casual de un síndrome de Gitelman. Med Fam SEMERGEN. 2014;40(7):95-8. https://doi.org/10.1016/j.semerg.2013.03.003
- Gil H, Mejía N, Alvárez-García O, Loredo V, Santos F. Longitudinal growth in chronic hypokalemic disorders. Pediatr Nephrol. 2010;25(4):733-7. https://doi.org/10.1007/s00467-009-1330-7
- Gómez de la F CL, Novoa JM, Caviedes N. Síndrome de Bartter: Una tubulopatía infrecuente de inicio antenatal. Andes Pediatrica. 2019;90(4):437-44. https://doi.org/10.32641/rchped.v90i4.932
- Knoers NV, Levtchenko EN. Gitelman síndrome. Orphanet J Rare Dis. 2008;22(3). https://doi.org/10.1186/1750-1172-3-22