
Vol. 28 - Num. 110
Notas clínicas
Síndromes vasovagales recurrentes en adolescentes: displasia arritmogénica de ventrículo derecho
Alberto Ibáñez Navarro a, M.ª Olaya Martínez Garbayob, Ana M.ª Izal Martínezb, Sonia Alfaro Virtoc, Silvia Calvo Pérezd
a, M.ª Olaya Martínez Garbayob, Ana M.ª Izal Martínezb, Sonia Alfaro Virtoc, Silvia Calvo Pérezd
aMédico de Familia. Consultorio Médico de Castejón. Castejón. Navarra. España.
bEnfermera Pediatría. Consultorio Médico de Castejón. Castejón. Navarra. España.
cEnfermera. Consultorio Médico de Castejón. Castejón. Navarra. España.
dEnfermera Pediatría. Hospital Reina Sofía. Tudela. Navarra. España.
Correspondencia: A Ibáñez . Correo electrónico: aibanez021@gmail.com
Cómo citar este artículo: Ibáñez Navarro A, Martínez Garbayo MO, Izal Martínez AM, Alfaro Virto S, Calvo Pérez S. Síndromes vasovagales recurrentes en adolescentes: displasia arritmogénica de ventrículo derecho . Rev Pediatr Aten Primaria. 2026;28:209-13. https://doi.org/10.60147/e17d4922
Publicado en Internet: 25-05-2026 - Número de visitas: 1192
Resumen
La displasia arritmógena de ventrículo derecho es una miocardiopatía infrecuente que puede producir arritmias malignas o incluso la muerte súbita en adolescentes debido al desconocimiento para su sospecha y a la dificultad para su diagnóstico. Puede simular un síndrome vasovagal con taquicardia sostenida, lo que lleva muchas veces a infravalorarla.
Palabras clave
● Arritmia ● Atleta ● Cardiomiopatía ● Desfibrilador ● Enfermedades de miocardioINTRODUCCIÓN
La identificación de cardiopatías en la población infantil sigue siendo un reto hoy en día en los servicios de urgencias, tanto rurales como urbanos, donde la atención sanitaria es generalmente por médicos generalistas, médicos de familia y/o urgenciólogos.
Existen alteraciones en los electrocardiogramas que muchas veces pasan desapercibidas a ojos no acostumbrados a su interpretación.
La displasia arritmogénica de ventrículo derecho es solo otro ejemplo más, ya que, aunque tiene una prevalencia muy baja ─algunos estudios la estiman 1:5000 and 1:2000─ puede conllevar un alto riesgo de muerte súbita por taquicardias de repetición.
El objetivo de este caso es ilustrar las características clínicas y diagnósticas de la DAVD en adolescentes deportistas, enfatizando su similitud inicial con síncopes vasovagales, para mejorar la sospecha diagnóstica en los servicios de urgencias y Atención Primaria.
CASO CLÍNICO
Varón de 14 años que acude al servicio de urgencias de un centro de salud rural por presentar episodio de pérdida de conocimiento brusco con relajación de esfínteres, mientras estaba jugando un partido de fútbol un día caluroso. La recuperación fue espontánea en menos de tres minutos.
Como antecedentes personales, no tomaba ningún medicamento, no tenía hábitos tóxicos y no tenía ninguna patología crónica conocida. Practicaba deporte con frecuencia, llevaba una dieta variada y no había presentado episodios similares. No tenía antecedentes familiares de interés.
Presentaba constantes normales, exploración física sin alteraciones significativas y se mantuvo en todo momento asintomático durante su estancia en urgencias, por lo que se dio el alta con observación domiciliaria.
Tres meses y medio después presentó un segundo episodio de menos de un minuto de duración, por lo que sus padres decidieron no consultar. Posteriormente, presentó un tercer episodio de síncope durante el ejercicio intenso, por lo que acudieron de nuevo al servicio de urgencias rural, donde se objetivó que la exploración, las constantes y el electrocardiograma eran normales (ritmo sinusal sin alteraciones de la repolarización). Debido a la presencia de síncopes de repetición en contexto de esfuerzo, se derivó al servicio de urgencias hospitalarias para valoración.
Durante su estancia en urgencias se mantuvo asintomático, la exploración física resultó normal y las pruebas complementarias no reflejaron hallazgos patológicos: la analítica sanguínea con hemograma, función renal, función hepática, ionograma sanguíneo y el estudio urinario fueron normales; el electrocardiograma con monitorización cardíaca con ritmo sinusal no presentó alteraciones electrocardiográficas reseñables, por lo que fue dado de alta con el diagnóstico de síncope vasovagal.
Una semana después presentó un nuevo episodio de síncope a los diez minutos de iniciado el ejercicio, de mayor duración, pues recuperó el conocimiento a los 6 minutos. De nuevo fue remitido a urgencias hospitalarias, donde, esta vez, dado el patrón recurrente de síncopes con esfuerzo, se sospechó cardiopatía estructural, motivo por el que se ingresó y se realizaron las siguientes pruebas complementarias: analítica sanguínea con bioquímica, hemograma, función renal-hepática, troponinas y sedimento urinario sin alteraciones. Electrocardiograma con ritmo sinusal a 55 lpm. QRS: 0,80 seg. Eje QRS 100°; extrasistolia ventricular con dos morfologías (una con bloqueo de rama izquierda y eje izquierdo y otra con bloqueo de rama derecha) y varios pares. Ondas T negativas en V1-4; micro-q en II, III y aVF. Radiografía de tórax: no cardiomegalia, aunque en proyección lateral se apreciaba crecimiento ventricular derecho. Vascularización pulmonar normal.
Se realizó un ecocardiograma transtorácico en el que se objetivaba ventrículo izquierdo (VI) no dilatado (DTD de 44 mm y DTS de 30 mm) con FEVI conservada (FE del 60%). Motilidad segmentaria normal. Ventrículo derecho (VD) dilatado con DFVD leve (TAPSE de 16 mm). Los valores normales en adolescentes se sitúan por encima de 17 mm. Ligera dilatación de AD. El resto del estudio, dentro de la normalidad (Figura 1). En la resonancia magnética cardiaca se observaba VI no dilatado con FVI conservada. VD dilatada 8 VTD 323 ml, VTS: 150 ml, con FE en torno al 36%. Hipoquinesia global de VD y adelgazamiento difuso de su pared. Movimiento paradójico de septo interventricular. AD dilatada (54 x 50 mm). Tras la administración de contraste iv no se observaron regiones de necrosis ni fibrosis.
| Figura 1. Síndromes vasovagales. Ecocardiograma transtorácico realizado |
|---|
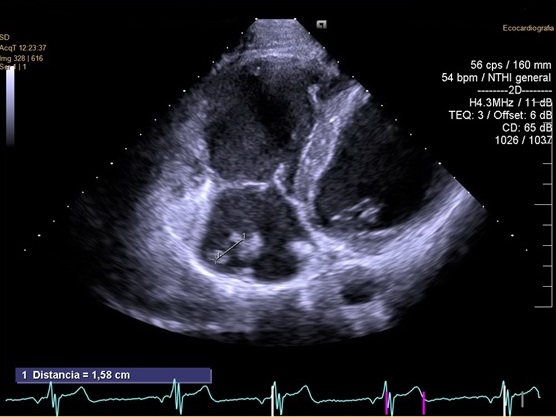 |

Se realizó una prueba de esfuerzo: fue submáxima 89% (188 l/min. 17,2 METs), clínicamente asintomático y sin cambios electrocardiográficos en la repolarización. Extrasistolia ventricular en bigeminismo y pares que desaparecían con esfuerzo y reaparecían en la recuperación. Progresión escasa de la TA de 20 mmHg con máximo esfuerzo.
En el holter de 48 horas se observó la presencia de extrasistolias ventriculares polimorfas muy frecuentes, con bigeminismo, pares y tripletes y una salva de 4 encadenados.
Con estos datos, se estableció el diagnóstico de displasia arritmogénica de ventrículo derecho (DAVD) según los criterios Task Force (Tabla 1), se inició tratamiento con atenolol, y se le dio el alta hospitalaria con la recomendación de suspender completamente las actividades deportivas.
| Tabla 1. Síndromes vasovagales. Criterios Task Force aplicados al caso clínico | |
|---|---|
| Criterios del Task Force de 2010 para diagnosticar la DAVD/MAVD | Caso clínico |
| 1. Disfunción global/regional y alteraciones estructurales (en RMN cardiaca) | Mayor: dilatación severa del VD o fracción de eyección ≤40% |
| 2. Caracterización tisular (biopsia) | No se realizó |
| 3. Anomalías de la repolarización (ECG) | Menor: ondas T invertidas en V1 y V2; o en V4-V6; o en cara inferior (II, III, aVF) |
| 4. Anomalías de la despolarización/conducción (ECG/holter) | Menor: ECG con potenciales tardíos o duración del QRS prolongada en el VD |
| 5. Arritmias | No cumple |
| 6. Historia familiar y genética | No cumple |
| Diagnóstico definitivo | 1 mayor + 2 menores |
| DAVD/MAVD: displasia/miocardiopatía arritmogénica del ventrículo derecho. | |

En posteriores revisiones en las consultas de Cardiología Pediátrica, siempre permaneció asintomático, no presentó nuevos episodios y había abandonado el deporte completamente.
Dada la alta carga arrítmica en los holter de control (bigeminismo casi constante, múltiples tripletes), presencia de síncopes previos y FE del VD del 36% en resonancia (persistiendo la hipoquinesia global en la ecografía), se decidió implantar desfibrilador automático implantable (DAI) según criterios de riesgo. Tras varios años de seguimiento, permanece asintomático sin activaciones del DAI. Se permite el ejercicio de moderada intensidad.
DISCUSIÓN
La DAVD es una enfermedad del miocardio caracterizada por atrofia muscular y el reemplazo del músculo por tejido adiposo o fibroadiposo1. Se ha relacionado con un defecto genético de trasmisión autosómica dominante con penetrancia y expresividad variables2. Se estima que tiene una prevalencia de 1:2000 y 1:5000, es más frecuente en deportistas o atletas y con predisposición en varones. Es muy rara por debajo de los 12 años.
La clínica más frecuente es un cuadro sincopal bien tolerado de carácter inespecífico en contexto de esfuerzo y con recuperación espontánea3. El electrocardiograma basal es normal, aunque es frecuente que aparezca una prolongación de QRS y de la onda S (V1-3) y/o un bloqueo de rama derecha y, en casos avanzados, una onda épsilon o fibrilación ventricular4,5.
Aunque la prueba de referencia es la angiografía y biopsia endocárdica, se utilizan habitualmente en la práctica otras técnicas menos invasivas para su diagnóstico (ecocardiograma y resonancia magnética) y se va ampliando el estudio con más pruebas según los resultados6.
El ecocardiograma tiene mayor disponibilidad y no es invasivo; por ello, es la primera prueba en realizarse para detectar áreas de discinesia, dilataciones y aneurismas. La resonancia permite, con gran sensibilidad, identificar el tejido adiposo del miocardio7. Otras pruebas de segundo orden, según los resultados de las pruebas anteriores, son la ventriculografía isotópica, la tomografía computarizada y el estudio electrofisiológico. Debido a su complejidad, se desarrolló una escala específica (Task Force Criteria reviewed)8 según la cual el paciente debe cumplir unos criterios mayores y menores para realizar el diagnóstico.
El tratamiento antiarrítmico se realiza con sotalol9 o amiodarona que pueden reducir las taquicardias sintomáticas, aunque no sustituyen la necesidad de implantar un desfibrilador en caso de mal control o alto riesgo de muerte; este último se puede estimar mediante la escala Calculadora de riesgos ARVC (ARVC risk calculator)10. En este caso, se prefirió utilizar la alternativa del atenolol como betabloqueante de primera línea. La ablación con catéter también puede ser útil en estos pacientes. En casos raros, también se contempla el trasplante cardiaco11.
CONCLUSIÓN
Casos como el descrito, con una clínica inespecífica y exploración inicial normal, recalcan la importancia de recopilar una historia clínica detallada, prestando especial atención a síntomas de alarma, como son los síncopes durante el esfuerzo, la recurrencia de los mismos y la prolongada inconsciencia, que obliga a descartar una cardiopatía estructural en los pacientes que los presentan.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Todos los autores han contribuido de forma equivalente en la elaboración del manuscrito publicado.
Los autores declaran que han obtenido el consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
ARVC: ARVC risk calculator · DAVD: displasia arritmogénica de ventrículo derecho · VD: ventrículo derecho · VI: ventrículo izquierdo.
BIBLIOGRAFIA
- Elias Neto J, Tonet J, Frank R, Fontaine G. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D) - What We Have Learned after 40 Years of the Diagnosis of This Clinical Entity. Arq Bras Cardiol. 2019;112(1):91-103. https://org/10.5935/abc.20180266
- Ackerman MJ, Giudicessi JR. Pediatric-Onset Arrhythmogenic Cardiomyopathy: Look Right, Look Left, Look Both Ways. J Am Coll Cardiol. 2019;74(3):359-61. https://org/10.1016/j.jacc.2019.05.023
- Cohen MI, Atkins MB. Arrhythmogenic right ventricular cardiomyopathy in the pediatric population. Curr Opin Cardiol. 2022;37(1):99-108. https://org/10.1097/HCO.0000000000000937
- Sevinç Şengül F, Tunca Şahin G, Özgür S, Kafalı HC, Akıncı O. Clinical features and arrhythmic complications of patients with pediatric-onset arrhythmogenic right ventricular dysplasia. Anatol J Cardiol. 2019;22(2):60-7. https://org/10.14744/AnatolJCardiol.2019.56985
- Te Riele ASJM, James CA, Sawant AC, Bhonsale A, Groeneweg JA, Mast TP, et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population: Clinical Characterization and Comparison With Adult-Onset Disease. JACC Clin Electrophysiol. 2015;1(6):551-60. https://org/10.1016/j.jacep.2015.08.004
- Te Riele AS, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: clinical challenges in a changing disease spectrum. Trends Cardiovasc Med. 2015;25(3):191-8. https://doi.org/10.1007/s00392-009-0751-4
- Gonzalez Corcia MC, Motonaga KS. Pediatric Arrhythmogenic Right Ventricular Cardiomyopathy: They May Be Small, But They Pack a Big Punch. JACC Clin Electrophysiol. 2022;8(3):319-21. https://org/10.1016/j.jacep.2021.09.014
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121(13):1533-41. https://doi.org/10.1161/CIRCULATIONAHA.108.840827
- Roudijk RW, Verheul l, Bosman LP, Bourfiss M, Breur JMPJ, Slieker MG, et al. Clinical Characteristics and Follow-Up of Pediatric-Onset Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Clin Electrophysiol. 2022;8(3):306-18. https://org/10.1016/j.jacep.2021.09.001
- Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA. ESC Scientific Document Group. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43(40):3997-4126. https://doi.org/10.1093/eurheartj/ehac262
- Orgeron GM, Crosson JE. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiol Young. 2017;27(S1):S57-S61. https://doi.org/10.1017/S1047951116002249